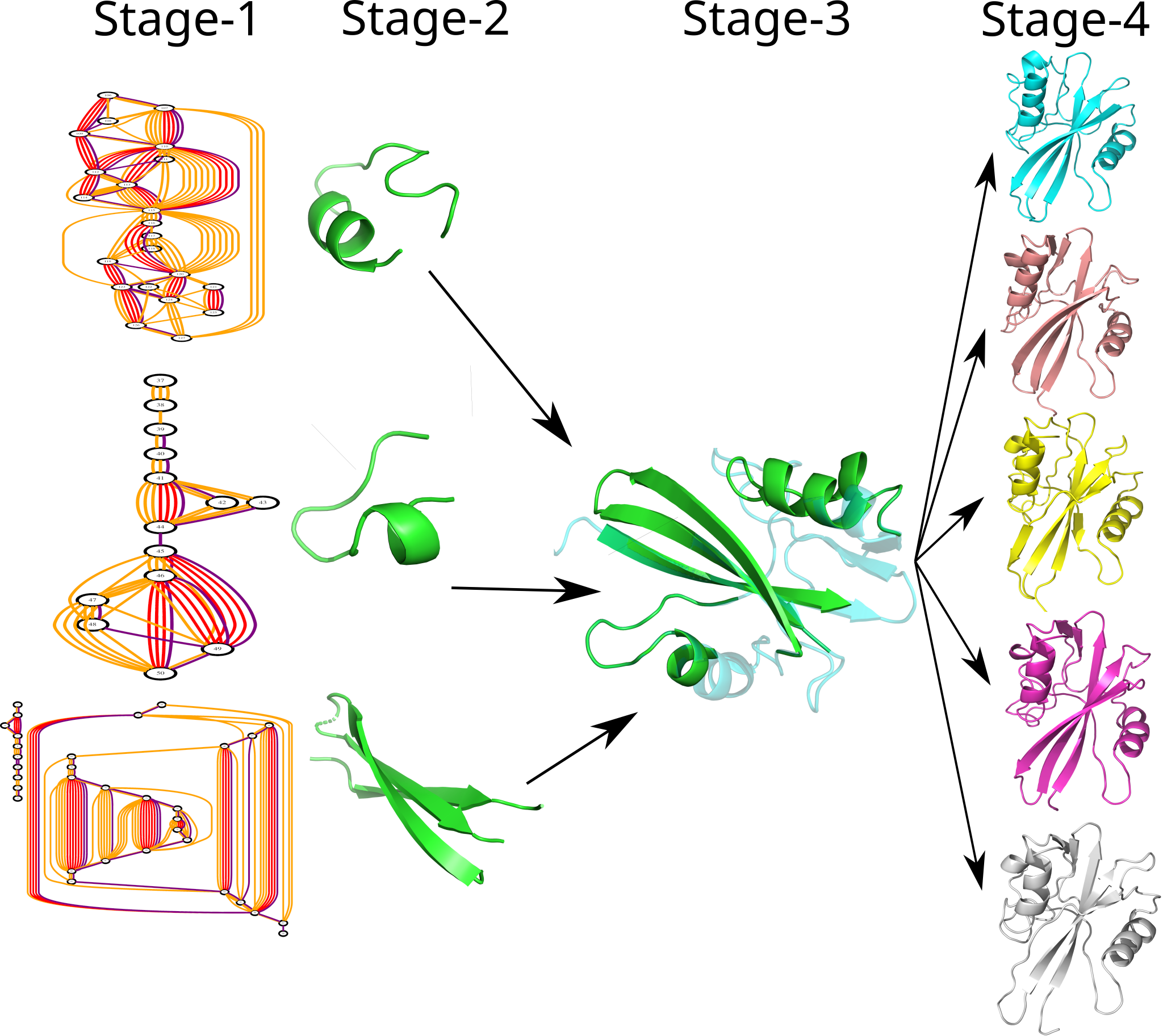 DREAM accepts the amino acid sequence, distance bounds, and dihedral bounds (if available) as input. The experimental bounds are typically distributed across regions of relative abundance and paucity. DREAM models the data in the form of a graph such that regions of relative abundance form core substructures that appears as dense subgraphs. The core substructures are modeled separately and in parallel in a distance geometric framework, before consolidating them in a single step. The regions of paucity are modeled separately leading to an ensemble of structures.
The native structure thus derived is dominantly driven by the experimental data. In order to model a structrue which is thermodynamically stable in solvent (water), energy minimization is carried only in the post processing stages to provide the final structure.
DREAM accepts the amino acid sequence, distance bounds, and dihedral bounds (if available) as input. The experimental bounds are typically distributed across regions of relative abundance and paucity. DREAM models the data in the form of a graph such that regions of relative abundance form core substructures that appears as dense subgraphs. The core substructures are modeled separately and in parallel in a distance geometric framework, before consolidating them in a single step. The regions of paucity are modeled separately leading to an ensemble of structures.
The native structure thus derived is dominantly driven by the experimental data. In order to model a structrue which is thermodynamically stable in solvent (water), energy minimization is carried only in the post processing stages to provide the final structure.
| Stage1 |
Stage2 |
Stage3 |
Stage4 |
- Read the input
- Model in the form of a graph
- find clusters with relatively larger
concentration of experimental
bounds.
|
- Model three dimensional
structure for the fragments
|
- Consolidate the structures
for the fragments
- model structures for the
regions with comparatively
less number of bounds
|
- Post process to get
a thermodymically
stable structure in
solvent (water)
|