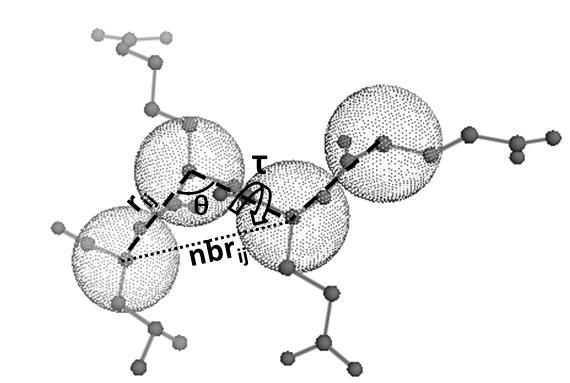
CGMM
Coarse Grained Molecular Mechanics forcefield for protein dynamics